Faculty of Chemistry, University of Belgrade, 11001 Belgrade, P.O.B. 158, Yugoslavia
Summary: - The mechanism of phenylselenoetherification of unsaturated alcohols involving
seleniranium cationic intermediates, is studied by semiempirical molecular-orbital MNDO-PM3
method.7-9 Results of calculation on several 4-alkenols attacked by PhSe+, reveal that the
reaction should be highly regio- and stereospecific.
INTRODUCTION
Intramolecular cyclization of unsaturated alcohols to cyclic phenylselenoethers (termed
phenylselenoetherification) involving a rearrangement of phenylseleniranium intermediate has
become an important tool for the synthesis of different natural products.1 At low temperature 4-
and 5-alkenols are converted into cyclic phenylselenoethers in good to excellent yields,
depending on the structure of the substrate and reagent used.2,3 It was shown experimentally that
4-penten-1-ol with phenylselenium chloride gives tetrahydrofuran ether (4a), whereas terminally
disubstituted alkenol (1d) gives a tetrahydropyran product (3d), in very good yields. The
reactions of monosubstituted 4-alkenols ((Z)- and (E)-4-hexen-1-ols) with phenylselenenyl
chloride show high regioselectivity. By the variation of the substitution on the 4 double bond,
the different cyclic ethers were obtained. The phenylselenoetherification of (E)-4-hexen-1-ol
gives the mixture of (Z)- and (E)-2-methyl-3-phenylselenenyltetrahydropyrans. Under the same
conditions (E)-4-hexen-1-ol (1c) yields in tetrahydrofuran ethers (4c and 4d). In this work we
present the results of the study involving cationic intermediate species, aiming to get detailed
mechanistic explanation of the experimental results. In the available literature there is no report
that this mechanism has been studied by semiempirical MO methods.
METHOD OF CALCULATION
The structures of compounds were generated by PC MODEL, version 4.0,4 that involves an MMX force field5 which was supplemented with parameters for selenium6 and were saved as MOPAC 7 files for MNDO-PM3 semiempirical calculations.8
In our work we used the MNDO method that proved to be highly reliable for investigating
molecular properties of molecules and ions.7-9 The MNDO-PM3 version is the only one
parametrized for selenium compounds.10 We used the MOPAC program package, Version 7.01.
The geometries of all molecular species correspond to the energy minima in a vacuum and were
optimized by the PM3 method. The transition states for all the reactions were found using
corresponding MOPAC facilities (TS, SADDLE). When needed, obtained structures were
refined by Bartel's method (Non-Linear Least Squares gradient minimization routine - NLLSQ),
and further proved by vibrational analysis showing only one negative vibration. The influence of
the solvent to the cations was not studied, because all the studied reactions were experimentally
done in nonpolar solvent.
RESULTS AND DISCUSSION
The general outline of the reaction investigated is given in Scheme 1. To make the most detailed correlation of calculations with experimental results we studied a homologous series of 4-alkenols derived from 4-penten-1-ol, having different substitution at position 5.
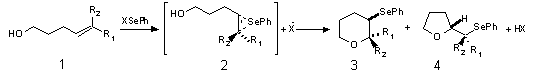
a: R1=H, R2=H
b: R1=CH3, R2=H
c: R1=H, R2=CH3
d: R1=CH3, R2=CH3
SCHEME 1
We have done the extensive calculations, by means of MNDO-PM3 method, including starting compounds, all the products, and cationic intermediates, given in Scheme 1, as well as corresponding transition states.
A phenylselenonium cation (PhSe+) is the reactive species that attacks the double bond in unsaturated alcohols giving cyclization products. Calculation of a reaction trajectory for the attack of PhSe+ on alkenol does not reveal any transition state. (Since the formation of seleniranium ion from selenyl cation and alkene is an exotermic process, the absence of any activation energy is expected.) Approach of two moieties proceeds smoothly giving the seleniranium cation intermediates 2 (Fig. 1). The cations of this type have already been reported in literature.11 The common feature of all the cationic adducts is that they possess seleniranium ring having two C-Se bonds of almost equal length (difference does not exceed 4%). Heats of formation of bridged cations 2a-d are given in Table 1.
| Table 1. Heats of formation of seleniranium ions (kcal/mol) | |||
| 2a | 2b | 2c | 2d |
| 139.7 | 124.3 | 126.1 | 114.0 |
Our calculations show that the intermediate cations 2 can be easily transformed into the six-membered cyclic ethers 3, bearing the phenylseleno substituent in position 3, and into five-membered cyclic ethers 4, bearing methyl-, 1-ethyl- or 2-isopropil-phenylselenenyl in position 2.
|
Table 2. Heats of formation of cyclic ethers (kcal/mol)* | |||
|
3a 3b
3c
3d |
42.4 eq 43.5 ax 47.9 tde 49.7 tda 50.9 see 49.5 sea 55.7 eq 54.5 ax |
4a 4b
4c
4d |
40.7 53.9 erythro
52.5 threo
61.8 |
|
* ax = 3-PhSe axial; eq = 3-PhSe equatorial; tda = 2-Me-3-PhSe erythro-diaxial; tde = 2-Me-3-PhSe erythro-diequatorial; see = 3-PhSe equatorial, 2-Me axial; sea = 3-PhSe axial, 2-Me equatorial. | |||
Products 3 and 4 have many stereo isomers. Structures in series a and d have one chiral center leading to two enantiomers. Products in series b and c have two chiral centers. Isomers labeled as b have an erythro configuration, and those labeled c have a threo configuration. Products 3b and 3c may have axially or equatorially oriented PhSe- groups. From the data in Table 2 may be seen that differences in the stabilities of axial and equatorial conformers are small compared with the activation energies for cyclization. (The axial preference of SePh groups in 3a and 3b should not be considered as a computational artefact, because it was found experimentally in some cyclohexane derivatives.12)
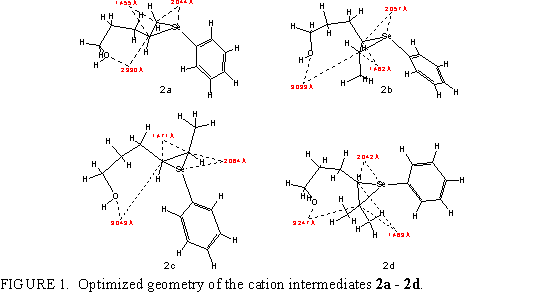
In Fig. 2 (left) the energy profiles are given for the cyclization of intermediate 2a to two most easily attainable ethers. Cyclization of 2a proceeds most smoothly to 4ap protonated on oxygen, via the transition state (pathway B) having the energy of 150.6 kcal/mol (activation energy 10.9 kcal/mol). It can be seen that this cyclization to five-membered cyclic ether goes through transition state of lower energy regarding the pathway B toward a six-membered ether (transition state of A has the energy of 155.6 kcal/mol). The difference in activation energy is 5.1 kcal/mol. This can account for the preferential formation of tetrahydrofuran products despite greater stability of the (non-protonated) tetrahydropyran products (see Table 2). Incidentally, the regioselectivity conforms to the Markovnikow rule, and to Baldwin rule via a 5-exo-trig mode.13 Experimentally, 5-membered cyclic ether, 4a, is found as almost the single product of cyclization.2, 3b
From the Figure 2 it may be seen that the energy of protonated cyclic ether is very close to the energy of corresponding transition state (only 2.1 kcal/mol lower). On the right side of Fig. 2 the transition states for the possible cyclization paths of 2a are shown. The common features are: C4C5 bond shorter than 1.5 Å, and CO bond very long, 1.9 to 2 Å. The structure of the transition state is not much different from that of protonated ether.
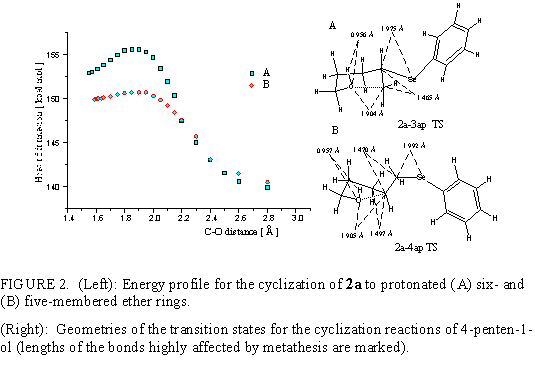
In experiments2 was found that the cyclization of 2d yields exclusively the tetrahydropyran product, 3d. This conforms to Markovnikow rule, and implying 6-endo-trig mode in Baldwin rule. Our calculations, depicted in Fig. 3, show that cyclization of 2d to 3d with equatorially oriented PhSe group has the lowest energy transition state. However, the corresponding cyclic ether (with equatorial SePh group) is not much more stable (less than 1 kcal/mol). The formation of tetrahydropyran products can be explained by conformation flip to a more stable cyclic conformer (with axial SePh group), and/or by the proton migration to selenium (more easily attainable in tetrahydropyran than in tetrahydrofuran products), giving a more stable protonated species.
The heats of formations for all the protonated ethers examined, are given in Table 3. (Suffix p
stands for protonated species.)
| Table 3. Heats of formation of intermediate
protonated cyclic ethers (kcal/mol).* | ||||
|
3ap
3bp
3cp
3dp |
150.1 eq 144.2 ax 138.5 tde 134.2 tda 140.7 see 139.4 sea 129.4 eq 124.2 ax |
4ap
4bp
4cp
4dp |
148.5 133.4 erythro
132.4 threo
128.9 | |
|
* See footnote in Table 2. | ||||
Calculations reveal that Se-protonated isomers are more stable. Difference in the stability of alternatively protonated ethers drops with increasing substitution at C-2. The activation barrier for the proton migration from oxygen to selenium in protonated 3a equals only 3.2 kcal/mol. This suggests a rapid proton transition, enabling a further stabilization of cyclization products. The corresponding proton migration in a tetrahydrofuran product, 4a, faces a much higher activation barrier (6.5 kcal/mol).
A striking difference in the reactivity of isomers 1b and 1c was reported.2 Cyclization of alkenol 1b with phenylselenium chloride gives tetrahydropyran derivatives 3b and 3c in the ratio 6.7 : 1. Same reaction of 1c gives only tetrahydrofuran derivatives 4b and 4c in the ratio 20:1. Markovnikow rule cannot be applied to these double bonds having secondary carbons on both sides, and Baldwin rule13, with its preference for 5-membered rings, is of no use for the explanation of experimental results.
Our calculations confirm that the most convenient approach of OH group to seleniranium carbon s from the opposite side to the coordinated selenium, as is expected for SN2 substitutions. Starting from such a molecular arrangement (reactive ground state) the (E)-4-hexen-1-ol (1b), via intermediate 2b, yields erythro cyclic ethers, and (Z)-4-hexen-1-ol (1c), via 2c, yields threo cyclic ethers.
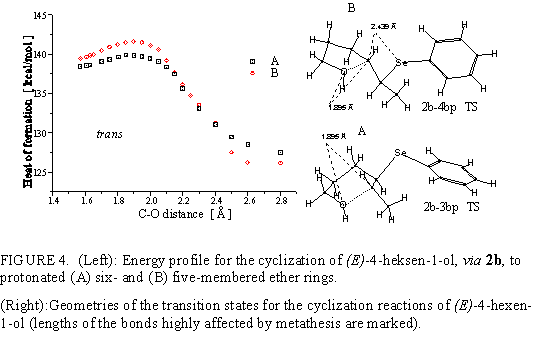
Calculated reaction coordinates for the cyclization of 2b are given in Figure 4. The transition
from 2b to threo tetrahydropyran, 3b, and tetrahydrofuran product, 4b, is energetically favorable.
The path to erythro products is more energy demanding, and obviously includes major
conformational change near the transition state. The reaction giving 3bp has the lowest energy
transition state (139.8 kcal/mol, activation energy 15.5 kcal/mol). Difference between transition
states leading to 3bp and to 4bp is ~1.8 kcal/mol. It corresponds to 6-fold faster formation of
tetrahydropyran derivative that can account for its preferential formation.
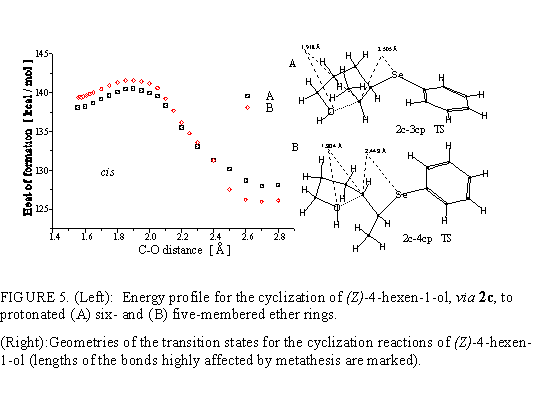
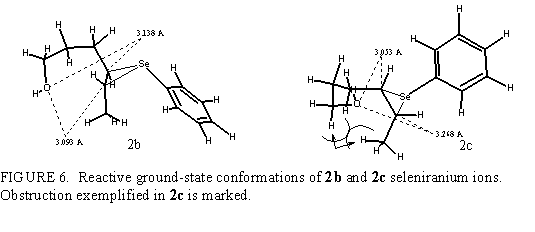
Discussion of the cyclization of 2c is highly analogous to that given for 2b. Corresponding reaction coordinates are given in Fig. 5. The preference for tetrahydrofuran products is obvious. Transition state that leads to 4cp has Hf=140.2 kcal/mol, i.e., the activation energy is 14.1 kcal/mol. Activation energy for the transition to 3cp amounts 18.2 kcal/mol, showing this path as inconvenient one. The obstruction due to interaction of terminal methyl group and C2 hydrogen (marked in Fig. 6) renders the approach of O toward C5 unfavorable.
CONCLUSIONS
Experimental results on the selenoetherification of compounds 2a-2d could be rationalized using Baldwin rule involving trigonal carbon atom undergoing ring-closure.13
Selenoetherifications of 4-penten-1-ol and 5-methyl-4-hexen-1-ol go on according to the Markovnikow and Baldwin rules, suggesting a carbocationic transition state.2 Our calculations show that the seleniranium cation is a stable intermediate. It comprises a strict nucleophilic attack on the seleniranuim ring carbon from the oposite side of the phenylselenil group.
Calculated regiospecificity of the addition of hydroxy and of a phenylseleno group in 4-penten-1-ol and in 5-methyl-4-hexen-1-ol matches the experimental findings.
In 4-hexen-1-ols there is no a priori preference for either side of double bond. The formation of six-membered cyclic ether demands only modest deformations of dihedral and bond angles. Such a stereochemical course is found in (E) alkenol. The same cyclization in (Z) isomer is rendered unfavorable by the repulsion (shown in Fig. 6) of terminal methyl group and CH2 group next to oxygen, which can be evaded by the formation of five-membered cyclic transition state.
Analysis of reverse process, the opening of cyclic ether, substituted with selenium at position 3, revealed that the protonation of cyclic selenoethers produces dramatic structural changes. One of the CO bonds lengthens to more than 1.55 Å. This elongation is accompanied with a ready approach of selenium 'from the rear' to the same carbon atom. Further bond elongation is promoted by stronger interaction with selenium, making the change in CO bond length up to 2 Å to be a very easy one.
From the Figures 2-5 could be seen that energies of protonated cyclic ethers are very close to
those of the corresponding transition states. Generally, the structure of the transition state is not
much different from that of protonated cyclic ether. Consequently, final stabilization of cyclic
ether should be coupled with the detachment of the proton from oxygen. It can be achieved by its
migration to a selenium atom in the same molecule, or to any other basic molecule in solution.
REFERENCES
(1) a) K. C. Nicolaou, R. L. Magolda, W. J. Sipio, W. E. Barnette, Z. Lysenko, M.M. Joullie, J. Am. Chem. Soc., 102, 3784 (1980); b) A. Krief, L. Hevesi, Organoselenium Chemistry I, Springer-Verlag, Berlin 1988; c) M. Tiecco, L. Testaferri, M. Tingoli, D. Bartoli, R. Balducci, J. Org. Chem., 55, 429 (1990); d) M. Tingoli, M. Tiecco, D. Chianelli, R. Balducci, A. Temperini, J. Org. Chem., 56, 6809 (1991); e) A. B. de Oliveira, D. S. Raslan, F. Khuong-Huu, Tetrahedron Lett., 31, 6873 (1990); f) N. Miyachi, H. Satoh, M. Shibasaki, J. Chem. Soc. Perkin Trans. 1, 2049 (1991); g) S. Patai, The Chemistry of Organic Selenium and Tellurium Compounds , Ed., Wiley, Chichester, 1986; h) C. Paulmier, Selenium Reagents and Intermediates in Organic Synthesis , Pergamon Press, Oxford, 1986; i) Liotta D, Organoselenium Chemistry , Ed., Wiley: New York, 1987; j) K. C. Nicolaou, N. A. Petasis Selenium in Natural Products Synthesis 'CIS' INC., Philadephia, 1984; k) P. Koovsky, M. Pour, J. Org. Chem. 55, 5580 (1990).
(2) a) S. Konstantinovi, Z. Bugari, S. Milosavljevi, G. Schroth, M. Lj. Mihailovi, Liebegs Ann. Chem., 261 (1992); b) S. Konstantinovi, Z. Bugari, R. Vukievi, W. Wisniewski, Z Ratkovi, Z. Markovi, M. Lj. Mihailovi, J. Serb. Chem. Soc., 62, 307 (1997).
(3) a) L. Clive, G. Chittattu, N. J. Curtis, W. A. Kiel, C. K. Wong, J. Chem. Soc., Chem. Commun.,725 (1977); b) K. Fujita, K. Murata, M. Iwaoka, S. Tomoda, J, Chem. Soc., Chem, Commun., 1995, 1641
(4) Serrene Software Box Bloomington IN 45402-3076
(5) a) J. J. Gajevski , K. E. Gilbert, J. McKelvey, Adv. Mol. Model., 2, 65 (1990); b) U. Burket, N. L. Allinger, Molecular Mechanics , American Chemical Society, Washington, DC 1982.
(6) a) N. L. Allinger, J. A. Allinger, L. Q. Yan, J. Mol. Struct. (Teochem), 201, 363 (1989); b) Z. Markovi, Lj. Doen-Miovi, I. Jurani, S. Konstantinovi, Ind. J. Chem., 34B, 695 (1995).
(7) a) J. J. P. Stewart, J. Comp.-Aided Molec. Des., 4, 1 (1990); b) J. J. P. Stewart, QCPE # 455.
(8) For PM3 parametrization of MNDO see: a) J.J.P. Stewart, J. Comp. Chem., 10, 109 (1989); b) J. J. P. Stewart, J. Comp. Chem., 10, 221 (1989).
(9) a) E. Bartoszak, Z. Degaszafran, M. Grundwaldwyspianska, M. Jaskolski, M. Szafran, J. Chem. Soc., Faraday Trans., 89, 2085 (1993); b) P. Burk, I. A. Koppel, Theor. Chim. Acta, 86, 417 (1993); c) H. Zuilhof, L. B. J. Vertegaal, A. Vandergen, G.Lodder, J. Org. Chem., 58, 2804 (1993); d) C. B. Aakeroy, J. Mol. Struct. (Teochem), 281, 259 (1993); e) M. W. Jurema, G. C. Shields, J. Comp. Chem., 14, 89 (1993).
(10) M. E. Amato, A. Grassi, K. J. Irgolic, G. C. Pappalardo, L. Radics, Organometallics, 12, 775 (1993).
(11) a) H. G. Schmid, D. G. Garratt, The Chemistry of Double-Bonded Functional Groups . Supplement A, Part 2, Chap. 9. Patai Ed., Wiley-Interscience, London; 1977; b) D. G. Garratt, H. G. Schmid, Can. J. Chem., 52, 1027 (1974).
(12) a) N. S. Zefirov, L. Gurvich, A. S. Shashkov, V. A. Smit, Zh. Org. Khim., 10, 1786 (1974); b) N. S. Zefirov, L. Gurvich, A. S. Shashkov, M. Z. Krimer, E. A. Vorobeva, Tetrahedron, 32, 1211 (1976); c) M. Mikolajczyk, P. P. Graczyk, M. W. Wieczorek, J. Org. Chem., 59, 1672 (1994).
(13) J. E. Baldwin, J. Chem. Soc., Chem. Comm., 734 (1976).
* Work supported in part by the Ministry of Science and Technology of the Republic of Serbia.
() To whom correspondence should be addressed.